
中国科大Nat. Chem.:MOF结构的动态扭曲促进光催化全水分解
利用太阳能直接将水分解为氢气,为缓解日益严峻的化石能源危机提供了一条切实可行的道路。但目前绝大部分的光催化水分解制氢反应都是牺牲剂存在条件下的半反应,能够直接实现全解水的光催化剂极其有限。实现光催化全解水的一个主要限制因素是光催化中激发态电子和空穴的快速复合。因此,抑制光催化过程中电子和空穴的复合成为实现光催化全解水的关键。为了抑制电子和空穴的复合,研究者们发展了许多有效的策略,但这些策略往往聚焦于催化剂的基态结构,而电子和空穴的复合却是发生在激发态。在自然光合作用中,光合蛋白中的各种辅因子孤立分布,保证形成高度局域的电荷分布。在接受电子后,辅因子通过结构变化实现对电子的稳定。受此启发,如果在人工光催化剂中可以允许电子供体和受体在光激发后形成高度局域的电荷分布,并且电子受体具有一定的结构柔性,那么便有望模拟植物体内的动态微环境,延长激发态电子的寿命,实现光催化全解水反应。
基于上述考虑,中国科学技术大学江海龙教授团队联合罗毅教授和江俊教授团队合作,基于一种名为CFA-Zn的金属有机框架材料(MOFs)实现了对上述过程的模拟。与传统光催化剂相比,MOF可以通过合理选择金属节点,实现对不同有机配体的分隔,创造电荷绝缘的电子供体和受体。同时,该MOF的有机配体具有一定的动力学结构(dynamic structure)特性,可以通过结构改变达到更优的能量状态,稳定激发态的电子。更重要的是,MOF具有明确的结构,可以从分子尺度上精确设计与调控,为进一步研究催化转化机制提供了极大的便利(图1a)。具体来说,CFA-Zn由Zn节点和两种化学上相同但晶体学上独立的有机配体组成,具有高度不对称的空间结构(图1b)。两种晶体学不同的有机配体分别作为电子供体和受体,而Zn的闭壳层结构保证了两种配体间的电荷绝缘。这些特征允许CFA-Zn创造类似植物体中柔性的微环境,实现对激发态电子的稳定,延长其寿命。此外,由于CFA-Zn微观结构上的不对称,其宏观上形成的不对称形貌可以进一步促进电荷迁移,从而促进水分解反应的进行(图1c)。
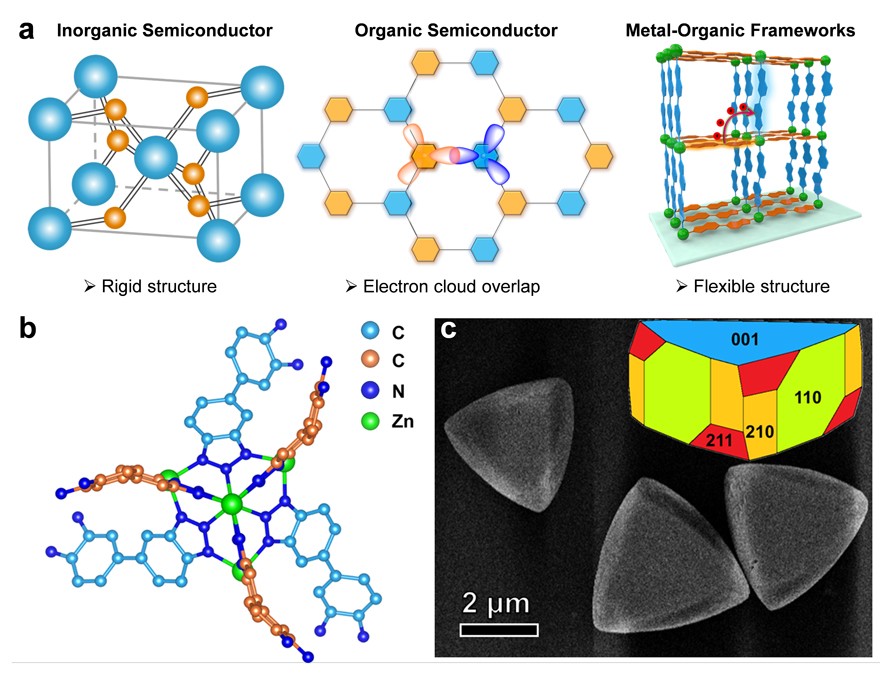
图1. (a) MOF实现激发态结构扭曲的优势;(b) CFA-Zn的次级结构单元及其连接了两种化学上相同而晶体学上不同的配体;(c) CFA-Zn的扫描电镜及模拟的形貌
首先,CFA-Zn的基态和激发态的轨道分布和结构分析为机制理解提供了重要的信息。CFA-Zn在基态时的VBM和CBM轨道分别位于两种晶体学不同的有机配体上,轨道在空间上高度分离(图2a,b)。由于VBM和CBM高度分离,具有CBM的有机配体在光激发后将得到激发态电子的全部能量,这会导致配体能量的急剧升高。为了实现对体系的稳定,配体通过结构变化达到更优的能量状态(图2c,d)。结构的改变会引起势能面的改变和轨道的重排。对于CFA-Zn来说,经过重排后其激发态的VBM和CBM轨道都位于相同的配体上(图2e,f),这意味着激发态的电子在由CBM弛豫回VBM的过程中仅发生在这一个配体上。而空穴依然留在基态时的VBM配体上(图2a),因此电子和空穴无法通过辐射弛豫过程进行复合,进而实现长寿命的电荷分离态,有望实现光催化全解水反应。
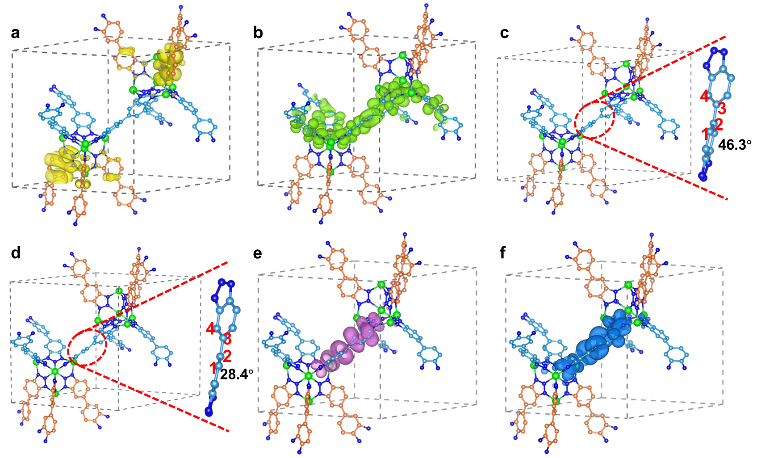
图2. CFA-Zn基态时的 (a) VBM和 (b) CBM轨道;CFA-Zn的 (c) 基态结构和 (d) 激发态结构;CFA-Zn激发态时的 (e) VBM和 (f) CBM轨道。
在上述理论分析的基础上,CFA-Zn光催化全解水的性能得到了实验证实。光催化中,通过光沉积引入Pt并外加Co3O4为共催化剂。从动力学曲线可以看出(图3a),反应初期有一个诱导期,这是由于Pt的光沉积过程引起的。随着反应时间的延长,H2和O2的比例逐渐偏离2:1,这是由于Pt共催化剂上发生的副反应和逆反应。CFA-Zn在365nm处的表观量子效率超过3%(图3b),这一数值是所有通过一步激发驱动全解水MOF中最高的。同位素标记实验证明,O2是来自于水分解,而不是空气的泄露(图3c)。CFA-Zn也具有良好的稳定性,连续反应100小时,活性仅有轻微衰减(图3d)。综合活性和稳定性,与其它经典的可见光一步激发进行全解水的催化剂相比也具有优势(图3e)。
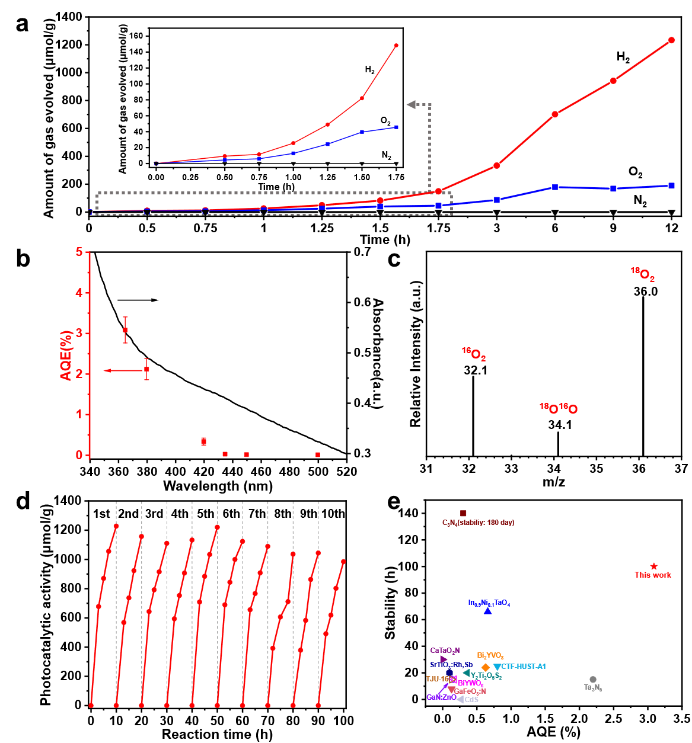
图3. Pt/CFA-Zn/Co3O4的(a)光催化动力学曲线和(b)表观量子效率随紫外可见吸收光谱变化结果;(c)确定O2来源的质谱测试;(d)光催化循环稳定性;(e)与其它经典可见光一步激发全解水光催化剂的性能对比。
CFA-Zn不对称的形貌可以进一步促进电子在不同晶面间的迁移,从而提升催化性能。理论计算表明光生电子大部分迁移到001晶面(图4a,b)。这意味着在光沉积Pt的过程中,Pt会优先在001晶面沉积,进一步增强MOF内建电场的强度,促进电荷的迁移。原位的表面光电压也从实验上证明了这一点(图4c,d)。光催化水分解是一个电子耦合质子转移的过程,质子迁移过程也十分关键。通过理论计算模拟质子在发生氧化(VBM)和还原(CBM)反应位点间的迁移过程发现,质子从氧化位点迁移到还原位点的能垒为0.719eV,这是一个比较低的能垒,容易克服。
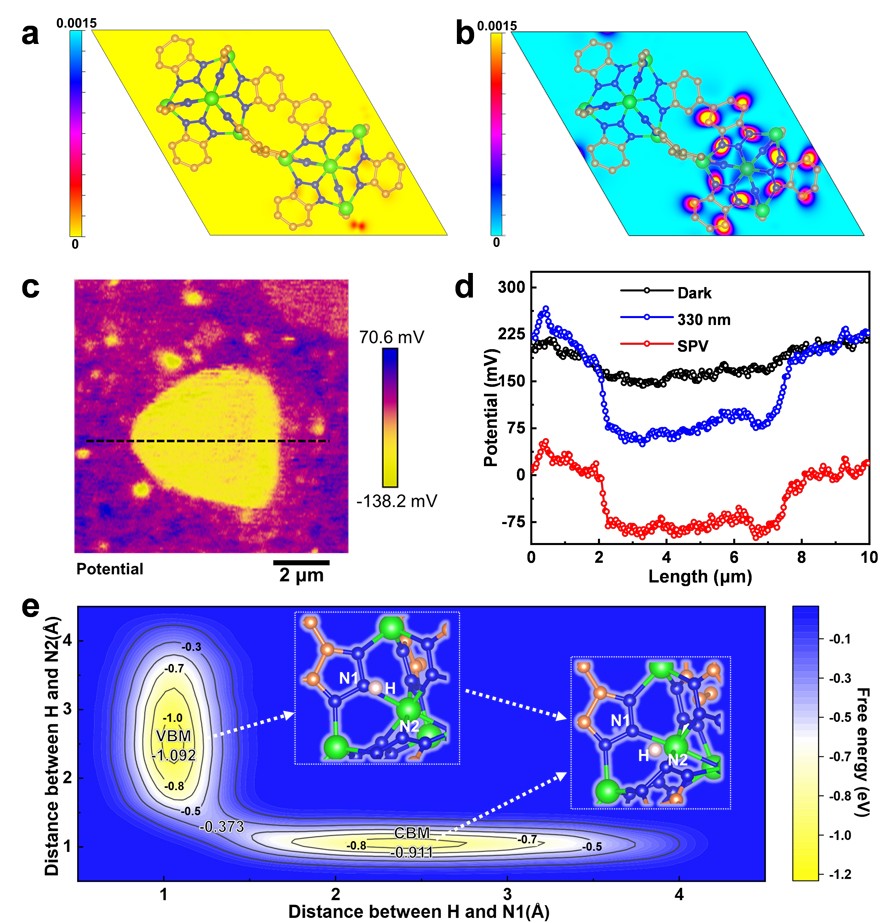
图4. CFA-Zn的(a) HOMO和(b) LUMO轨道在001晶面的投影;(c)原位表面光电压成像;(d)沿(c)图中黑色虚线的表面光电压;(e)对质子迁移过程的模拟。
为了支持激发态结构扭曲抑制辐射弛豫过程的机制,作者基于MOF易于结构修饰的特性对反应机制进行了验证。首先,在MOF的配体中引入具有空间位阻的-CH3官能团,直接抑制其激发态下的扭曲。计算结合光谱实验证明,-CH3的引入抑制了配体的旋转,导致电子和空穴的快速复合。光催化性能也随之下降。其次,通过在MOF中将Zn节点部分替换为Co,破坏其激发态高度局域的电荷结构。随着Co引入量的增加,光催化活性快速下降(图5a)。原位的XAS证明,Co参与了电荷的转移(图5b)。在这种情况下,激发态的电子被配体和Co节点共同分担,配体的能量改变不足以引起结构的扭曲(图5c)。动力学模拟和超快光谱也都表明,掺入Co以后,MOF的激发态电子迅速与空穴复合(图5d,e)。
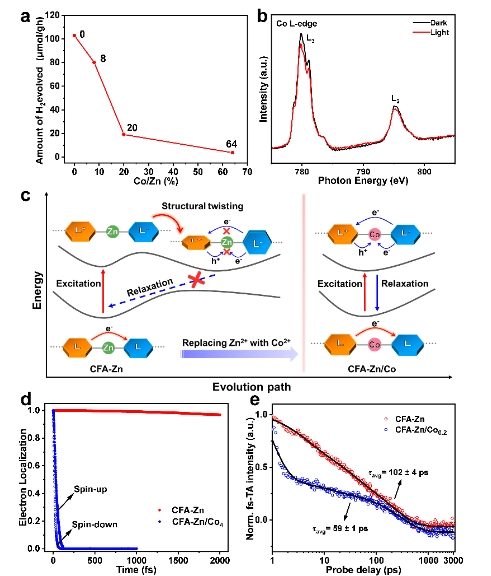
图5. (a)掺入不同量Co后MOF样品的光催化全水分解活性;(b)部分Zn节点被Co取代后样品CFA-Zn/Co0.64的原位Co L-边X射线吸收谱;(c)CFA-Zn通过结构扭曲抑制辐射弛豫过程,而引入部分Co之后无法通过结构扭曲抑制辐射弛豫的示意图;(d)对CFA-Zn和CFA-Zn/Co4激发态电子弛豫过程的理论模拟;(e)CFA-Zn和CFA-Zn/Co0.2的超快光谱。
相关研究成果发表在Nature Chemistry 上,该论文共同第一作者是中国科大博士后孙康和特任副研究员黄炎,江海龙教授和江俊教授为共同通讯作者。应期刊编辑邀请,研究团队、审稿人和编辑共同撰写的Research briefing也在线刊登。期刊审稿人和编辑对该工作给予高度评价,称该工作提出了一个“新颖、颠覆性的概念(novel and disruptive concept)”,并认为这为未来发展更高效的光催化剂提供了“令人激动的进展(exciting development)”。
